金币
UID25184
帖子
主题
积分221043
注册时间2012-4-1
最后登录1970-1-1
听众
性别保密
|
欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
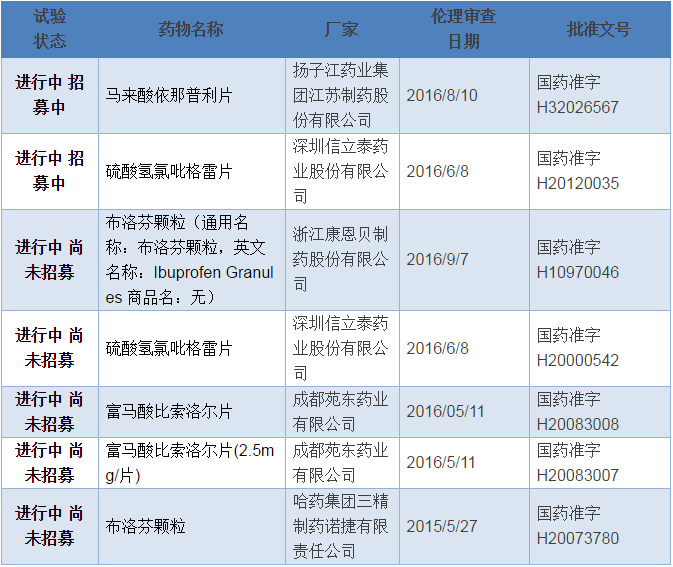
到2016年11月24日为止,在CDE临床试验平台备案的CFDA要求的289个品种一致性评价的项目仅有5个药品,7个厂家规格(文号),如下表。
按照一般的进程,从在 CDE临床备案到完成人体生物等效试验需一年左右,也就是说,到2017年底,有望完成SFDA要求的289个基本药物一致性评价仅有5个药品,7个厂家规格(文号)。
按照SFDA的要求,这289个药品的1万6千多个文号要在2018年前完成一致性评价,目前的进程表明,这不仅注定是一个落空的任务,也是不可能的任务,甚至是在2018年前还基本没有开始的任务。
影响了一致性评价的关键因素有哪些呢?一、艰难海淘的参比制剂。
到目前为止公布了三批个产品的参比制剂,这些制剂均是在海外注册生产和销售,除了购买不同批号外,还要艰苦收集相关合法和合格资质证明,这些与虎谋皮的行为注定了不会得到国外企业的配合,加大了一致性评价的难度,拖延了进程。
然而生产这些制剂的跨国公司在国内设立的中国公司子生产的相同的产品却不能当做参比制剂,同时,令人感到错愕的是这些国内子公司在国内地产化的产品在招标时却被全部当做原研药品对待享受了原研药的分组和价格。
这样两个环节的不同待遇被指就是一方面为了加大国内企业一致性评价的难度、一方面外企可继续保有市场优势。而一般原研药品由国内子公司生产时均是照搬国外的处方或是稍加改进,并不会有实质性变化。
11月22号SFDA公布的仿制药质量和疗效一致性评价工作政策问答中关于参比制剂选择问题:原研品种难以确定或已停产、退市,美日无相关RLD(reference list drug)产品。
SFDA答:按照《总局关于落实<国务院办公厅关于开展仿制药质量和疗效一致性评价的意见>有关事项的公告(2016年第106号)》的要求,企业找不到且无法确定参比制剂的,由药品生产企业开展临床有效性试验。
这一条更是匪夷所思,可能是混淆了一致性和有效性的区别,没有确定参比制剂的开展临床有效性评价,那么跟谁做?是和空白对照比有效就可以,还是要有一个参比制剂做非劣效试验?如果是前者,做有效性试验那就不是一致性评价,如果是后者还得需要一个参比制剂。
况且临床试验相比测定血药浓度的等效性试验可变因素太多,要判断起来要困难的多。
有些参比难道一定要病态的钻牛角尖式的追踪到数十乃至百年前的原研药品厂家?即使追踪到那个厂家,那么现在的辅料工艺也不是数十年前当时的药品的处方和工艺了,随着时代的进步也不断更新了设备工艺和处方了。
二、海量的生物等效性试验需求和紧缺的一期实验室生物等效试验能力
从CDE开通临床试验备案之日起,在其系统中的等效性备案共543个,计算下来每年接受生物等效试验不过一二百个,722临床核查后临床机构更趋向于少接慎接,除去新药和注册的生物等效试验外,能够进行一致性评价的寥寥无几,即使传说的放宽生物等效试验机构的认证或放开生物等效试验的取血,建立起一套能够经得起核查稽查乃至侦查的质量保证系统到进行试验至少也得一年以上,对2018前完成一致性评价完全无济于事,帮不上忙。
三、一致性评价决胜在市场
市场配套的政策才是企业主动进行一致性评价的最大动力,而现在看招标政策并不配套。
从一致性评价概念提出之日,跨国公司就赢定了,因为这从概念上确定了原研药的优势地位,原研药(包含其国内子公司地产化产品)在卫计委的招标规则中单独分组并不和国内企业比较,拉大了原研和国内企业的价格差,占尽先机国内企业望其项背而不能及。
企业要证明口服制剂的一致性只有重新进行生物等实验这华山一条道,以前的上市时的BE试验数据并不被承认。令人费解的是注射液等进行药学研究容易被证明一致性的剂型并不开展,也没有任何渠道得到招标部门的认可。
在招标规则中空有一条通过一致性评价的药品单独分组,一直没有任何一个产品能够运用这个规则,所以有人认为一致性评价客观上是帮了原研企业的忙,每年也也多消耗了数百亿的医保费用。一致性评价进展缓慢,外企才是一致性评价中最大赢家。
|
|


 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033





 发表于 2016-12-5 21:10:15
发表于 2016-12-5 21:10:15
 置顶卡
置顶卡 变色卡
变色卡

