引言:药品生产过程(CMC)时常发生各种变更,场地变更是其中一项重要变更,存在不同程度的风险。监管机构对变更申请的管理注重变更风险控制,以保证变更前后药品质量的一致性,美国自19世纪80年代就对药品生产变更进行了探索,本文将介绍相关法规规定。
本文针对已上市药品的生产变更问题,通过对美国化学药品生产变更立法历程和法规体系建设、药品生产变更报告类型的划分以及变更前后药品质量保证,并针对生产场地变更进行分类管理,并在此基础上探究美国是如何降低生产变更体系中政府以及企业的负担,从而为我国进行药品生产变更提供一些初步的建议。
美国化学药品生产场地变更的
法规研究
李晓宇 杨悦(通讯作者)
国际食品药品政策与法律研究中心
美国药品生产变更的立法溯源
美国已上市药品生产变更的相关法规的形成分为三个阶段。初始立法始于1982年,美国相关立法部门在21CFR第314.70章节中对生产变更做出了相关规定,并后续发布了一列配套指南(简称SUPAC系列指南)[1],此时,生产变更立法并没有上升到法案的级别,即在FDCA中并没有相关的规定;直到1997年,美国国会通过的FDA现代化法案(FDAAA)的第116章节对药品生产变更做出了规定,把生产变更上升到了美国药事法中的最高级别,即FDCA的第356a章;为了配合该法案实施,1999年,FDA相关部门拟定修订了1982年制定的21CFR第314.70章节,并发布了针对已批准NDA和ANDA的生产变更的比较全面的指南(Changes to an Approved NDA or ANDA),该指南对1999年之前发布的SUPAC系列指南相关内容进行了整合,但并没有完全替代。2004年,FDA通过搜集各界自1999年以来提交给FDA的提议,制定了21CFR第314.70章节最终法规,并对1999年发布的指南做出修订[2]。至此,美国的化学药品生产变更体系完全建立。
在上述法律体系中,FDA始终应用风险评估的方法对生产变更进行监管。在90年代,FDA与Maryland大学等发起一场研究,研究速释口服固体制剂的CMC变更对药品性能、安全性和有效性的影响。通过该研究,CDER确立了风险评估的方法[2]。1997年的现代化法案将风险评估列入生产变更之中,并授权FDA根据生产变更对药品存在的潜在风险性对生产变更进行监管。
在1982年至1997年间,FDA依据生产变更对药品的安全性和有效性存在的潜在影响将所提交变更报告分为三种:即事先需审评的补充申请(PAS)、立即生效的补充申请(即事先不需审评,CBE)以及年度报告(AR)。1997年之后,考虑到一些变更的风险性,以及加强对这些变更的风险控制,把立即生效的补充申请分为两类,即30天后生效的补充申请(CBE-30)和立即生效的补充申请(CBE)。
美国药品生产变更的分类
21CFR第314.70(a)(1)(i)条款规定,申请人对已批准的上市许可进行变更必须以相应报告的形式告知FDA。药品生产变更按事项分为以下几类:①对药品成分和组分(components and composition)的变更;②生产场地(manufacturing site)的变更;③生产工艺的变更;④药品规格(specification)的变更;⑤药品容器封闭系统的变更;⑥药品标签的变更;⑦其他变更以及⑧关联变更(multiple related changes)。
上述每一生产变更事项均包含不同的生产变更类型,不同生产变更类型对药品安全性和有效性的潜在影响程度不同,需进行不同程度的监管。因此,根据同一变更事项下的不同的生产变更类型对药品的安全性或有效性存在的潜在风险程度,美国法规划分了三种程度的变更,不同程度的变更提交监管程度不同的报告。潜在的风险程度表现为对影响药品的安全性或有效性因素,例如药品的均一性、规格、质量、纯度或效力存在潜在不良影响的程度。
表1不同程度生产变更的分类
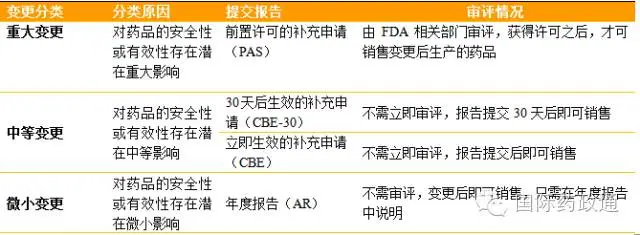
在表1中,重大变更对药品的安全性或有效性存在潜在影响最大,FDA对其所提交的PAS报告的监管力度也最高。对于中等变更,CBE-30报告的严格程度相对高于CBE报告,绝大部分该类生产变更需要提交CBE-30报告,只有少部分的非主要变更提交CBE报告。申请人提交CBE-30报告后,FDA会在30天内对该报告进行形式审查,以确定报告中的生产变更是否属于重大变更需要提交PAS报告,或审核信息的完整性,如果缺乏某些信息,会告知申请人补充相关资料。申请人收到FDA的回复后,就必须提交PAS报告或补充缺乏的信息,直至信息补充完备后,才可上市销售变更后生产的药品。然而,对于提交CBE报告的变更可在报告提交之后立即销售变更后生产的药品[3]。
FDA将企业内部与企业间的场地变更均视为场地变更,境内外场地变更管理一视同仁。根据风险程度划分需提交的申请类型。而我国对生产企业间和企业内部的生产场地变更实行不同的监管方式,境外生产场地变更和企业内部生产场地变更按照药品注册补充申请的要求进行监管,需提交补充申请,企业间的场地变更则视为委托生产,依据《药品委托生产监督管理规定》进行监管。
对生产场地变更的相关规定

一般性规定

美国对于生产场地变更的法规适用于申请人自身拥有或委托生产的场地变更,并且适用于国内外的生产场地变更[3]。
生产场地的变更,除了监管信息和场地信息变更外,不能涉及其他事项变更,例如扩大生产或生产工艺变更,并且标准操作程序(SOPs)、具有生产加工经验的人员、环境条件和质量控制,以及生产批次记录等也必须保持不变。但是,如果涉及其他事项变更,就视为关联变更,就必须按照单个变更中报告最为严格的类型提交。例如变更原料药生产商,该变更不仅涉及生产场地的变更,还会涉及诸如生产设备、生产工艺等的变更,申请人若有足够的信息评估原料药不同来源间的差异,就可视情况选择合适的报告类别,如果不能评估原料药或药品制剂不同来源间的差异,即必须提交按照单个变更所需提交的最高级别的报告。
生产地点变更的具体规定
在美国相关法规以及指南中,药品生产场地分为(1)药品、半成品(in-process materials)、原料药或原料药中间体(drug substance intermediates)的生产或加工(manufacturing or processing)场地;(2)首次或二次药品包装场地;(3)首次或二次药品贴标场地;(4)化学成分(component)、药品容器、密封材料(closures)、包装材料、半成品(in-process materials)、或药品制剂的检测场地以及进行稳定性试验的场地。
场地变更中,生产场地的cGMP检查状态、生产场地所进行的操作类型以及所生产药品的类型(例如原料药中间体、原料药、药品制剂)是对药品的安全性或有效性存在潜在影响的三个主要因素,三者融合贯穿于整个生产场地变更法规之中,对所要提交的报告类型起着决定性的作用。 CGMP检查状况对所提交的报告类型的影响
FDCA第351 (a) (2)(B)条款规定,药品的生产、加工、包装或储存过程中使用的生产方法、生产设施或过程控制,必须符合或者按照21CFR中对cGMP的要求操作,以确保药品的安全性,以及应具有的均一性、规格、质量、纯度,否则,该药品被认定为掺假药(adulterated drug)。因此,进行生产场地变更,申请人必须首先考虑新生产场地的cGMP检查状态。
不论是生产或加工场地,还是包装、贴标、检测场地(除了原料药中间体的生产场地),若其cGMP检查不合格,都必须提交PAS报告。对于转移到FDA从未检查过的生产场地(除了原料药中间体的生产场地),也必须提交PAS报告。
对于cGMP检查合格的新生产场地,要考虑cGMP检查距离变更的间隔时间长短。如果cGMP检查后,新生产场地未投入生产,不论该检查发生在2年之前还是2年之内,只需提交CBE-30报告,如果新生产场地是已批准的上市许可中写明的生产场地,就不必告知FDA;若新生产场地在检查过后投入生产,进行变更时,终止生产时间超过2年,就必须提交PAS报告,但若终止生产时间在2年之内,则提交CBE-30报告[3][4]。cGMP检查以2年为分水岭,是因为美国FDCA相关条款规定,对于已经经过cGMP检查的生产场地,在以后的生产过程中必须在连续2年内进行一次cGMP检查[5]。
上述分析中,把原料药中间体新生产场地的cGMP检查排除在外,并不意味着FDA不进行检查,而是不对其进行常规检查,实行“有因检查(cause inspection)”,即出现问题后,再对其进行检查,但是,如果该新生产场地cGMP检查不合格,也必须提交PAS报告[4]。
下文生产变更分析均是在cGMP检查合格的条件下进行分析
同一生产地点内的生产场地变更
美国相关指南对美国境内与境外的同一和不同生产场地的规定不同。在美国国内,同一生产地点是指拥有同一个场地注册时FDA给予的场地注册号码(establishment registration numbers)、并由同一个FDA地区办公室进行检查的新旧建筑物集合;不同生产地点是指,新旧建筑物拥有不同的场地注册号码,或者由不同的FDA地区办公室对其进行检查。在美国境外,国外同一生产场地是指新旧建筑物必须紧邻(adjacent)或毗邻(contiguous),之间间断或有间隔就视为不同生产场地。
除了表2中的特殊药品外,在同一个生产地点内进行生产建设活动,或在同一生产地点内的某一建筑物内或建筑物间转移生产操作,不必告知FDA。
表2同一生产地点内的生产场地变更的特殊情况

不同生产地点之间的生产场地的变更
药品制剂的完整生产流程中,美国比较注重生产或加工场地、首次包装场地变更的影响,因为该类变更对于各类药品安全性或有效性或所进行的生产操作产生的会直接产生很大的影响,且不确定性程度高,因此,所提交的报告类型相对较高级;二次包装场地、贴标以及检测场地对于各类药品安全性或有效性或对所进行的生产操作关联性不大,所需提交的报告类型相对低级,而且所有的类型的药品所提交的报告类型相同。
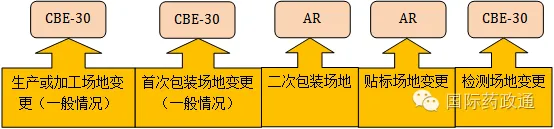
图1生产场地变更所需提交的基本报告类型
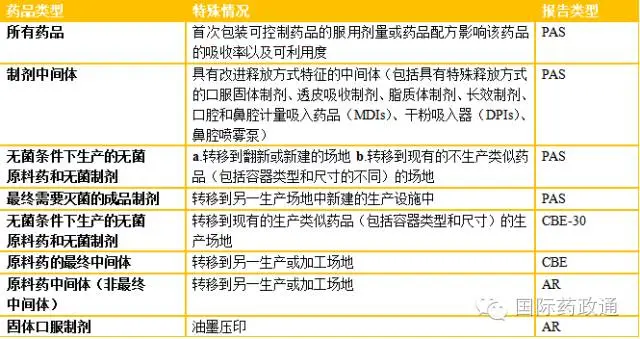
生产或加工生产场地的变更,除了特殊情况,其他所有原料药、制剂中间体以及药品制剂均只需提交CBE-30报告;以下表中的所列举的情况必须根据相关规定提交相应的报告:
表3不同生产地点之间的生产场地变更的
特殊情况
其他场地变更
对于包装材料以及辅料的生产场地的变更,美国相关法规或指南中没有详细的规定。但是,包装材料的灭菌过程若与已批上市申请中的灭菌过程有本质上的不同,需要提交AR报告;辅料的生产场地变更可不必告知FDA [4]。
生产场地变更后的药品质量保证
FDA依据生产变更的潜在风险性从立法源头、药品生产过程控制以及事后结果验证三方面来保证药品生产场地变更后药品的安全性和有效性。
立法之时,FDA是根据生产变更对药品的安全性和有效性存在的潜在影响规定报告类型,而非生产变更前后的实际验证结果。这使FDA在操作中占据主动性,防止危害药品安全性和有效性的生产变更的发生。FDA相关部门通过审查提交的报告,可确定申请人是否提交了正确的报告类型,生产变更是否对药品的安全性与有效性存在不良影响,以及影响程度。这种方式虽然在某种程度上削弱了生产企业自主决定生产变更报告类型的主动性,但是,可从根源上保证变更前后药品的安全与有效,防止危害公众健康的药品的销售。
场地变更之时,新场地的cGMP检查状态是申请人必须首先考虑的因素。如果场地的cGMP检查合格,那么,申请人就可根据相关规定提交FDA所建议的报告类型,但是,如果cGMP检查不合格,那么,就必须提交最高级别的PAS报告。申请人可从FDA内部的自由信息办公室(Freedom of Information Office,FOI)获得有关新生产场地的cGMP检查状态的信息,该办公室会向申请人发送一份质量保证文件(Quality Assurance Profile,QAP)的复印件作为凭证。cGMP检查可保证药品生产过程中的合规性。
场地变更后,申请人必须依照相关规定对变更后的药品的安全性和有效性进行评估。美国FDCA相关条款规定无论是重大变更、中等变更或是微小变更,在销售变更后生产的药品之前,申请人必须验证(validate)/评估(assess)变更对影响药品安全性或有效性的因素,例如药品的均一性、规格、质量、纯度或效力的影响[6]。
评估场地变更后药品的安全有效,主要表现在验证药品质量各项重要指标是否达到相应标准。生产场地变更所涉及的原料药中间体、原料药、制剂中间体或药品制剂的各项质量指标必须符合已批准的上市许可中制定的质量标准(specification),即利用相应分析程序(Analytical Procedures)对变更后生产的产品进行检测(Additional Testing),包括化学、物理、微生物、生物检验、生物利用度试验以及/或稳定性试验,其检测结果符合规定的验收标准(acceptance criteria)。这些检测试验并不是全部进行,要依据药品生产变更的类型、药品或原料药的类型以及变更对药品质量的影响程度而定。例如,对于超出已批准的上市许可范围内的生产场地的变更,除了进行基本的检测之外,还需进行生物等效性试验[7]。所采用的检测方法和研究试验,以及产生的相关研究数据必须真实地包含在所提交的补充申请或年度报告中。
赋予FDA和生产企业灵活性
减轻各自的负担
生产变更复杂多样,然而监管资源有限,如何平衡这两方面之间的关系?美国国会在1995年通过了削减文书工作法案[Paperwork Reduction Act of 1995 (44 U.S.C. 3501-3520],用以指导美国政府在制定各种法规之时,减少政府以及相关企业的文书负担。从生产变更法规的制定到实行,FDA始终贯彻上述法案的理念,在保证药品质量的前提下积极降低FDA的监管负担,以及企业的负担。
应用风险评估的方法减轻FDA的
监管负担
在立法之初,FDA就将风险评估的方法应用到生产变更之中。该方法使FDA对不同类型的生产场地变更实行不同程度的监管,降低FDA对低风险的生产变更的资源投入,加强对高风险药品生产变更的监管,从而有效分配有限的监管资源。
通过对生产变更的风险性进行评估,FDA积极降低已证明低风险性的生产变更所提交的报告级别。随着生产技术的创新或者新信息的发现,或根据以往的审评经验认为某些变更的风险性降低,FDA就会降低这些生产变更所提交的报告级别。这种方式可减少提交的补充申请的数量,而不会增加年度报告的数量,从而降低FDA审评补充申请的负担。在2014年发布的指南中,FDA根据以往的审评经验,把一些曾列为提交补充申请的重大变更认定为微小变更。例如,无菌制剂以及无菌原料药的生产场地的变更,在以往的指南中通常提交PAS报告或CBE-30报告,但是在该指南中,在现有的传统灌装场地内的灌装或合成区域内增加隔离板(barriers)以减少常规人员操作,或对生产设施进行微小结构修改(minor structural modifications)就不必提交PAS报告,可在年度报告中提交。[8]
另外,采用风险评估对生产变更进行监管,可激励生产企业进行生产技术创新以及加强对药品质量控制系统的控制。对于生产企业来说,药品上市时间的早晚对企业利润会产生很大的影响,特别是需提交PAS报告的重大生产变更。美国FDCA相关条款规定,补充申请除了所提交的材料与上市申请侧重点不同外,其审评过程与上市申请相同,严格的审评意味着审评时间的延长,这对变更后生产的药品的上市极为不利。因此,生产企业积极对生产技术进行创新,使药品生产获得更加安全的保障,从而降级变更报告级别,争取缩短药品上市时间。这种方式也在一定程度上降低了企业自身的变更申请负担,同时,也减轻了FDA的监管负担。

运用可比性研究计划减轻企业进行
生产变更的负担

为了降低生产企业提交生产变更报告的负担,尤其是提交PAS报告的负担,生产企业可通过向FDA提交可比性研究计划(comparability protocols)降低生产变更的风险性,从而降低报告级别[9]。可比性研究计划可预先提供将来进行生产变更需要进行的试验和研究、分析程序以及评估生产变更影响的验收标准。完备的可比性研究计划可为FDA提供充足的信息,供其事先评估生产变更是否存在潜在不良影响,并决定是否降低该生产变更的报告类型。通常情况下,可比性研究计划范围内的生产变更的报告级别低于没有提交可比性研究计划的生产变更的报告级别(例如从PAS降为 CBE-30, CBE, 或 AR)。很多情况下,可越过多个等级(例如PAS 直接变为AR)。
可比性研究计划必须在生产变更之前提交,或与上市申请一起提交。获得FDA审评后,申请人在进行生产变更时,就可使用研究计划变更中的试验和研究数据等信息,而不需重复进行试验或研究。这对于需要重复进行的生产变更来说,十分有用。对于生产场地的变更,申请人必须在协定中事先声明新生产场地的cGMP检查合格。
另外,对于事先需审评的补充申请,21CFR314.70规定,出于对公众健康的原因,或补充申请中变更的延迟将会给申请人增加特殊的困难,申请人可要求FDA加速审评事先需审评的补充申请,得到加速审评的PAS只需4个月就可完成审评,相比于正常情况下的PAS来说缩短了2个月[10]。
FDA采取的上述措施取得了一定的效果,有效抑制了PAS的数量的增长,减轻了FDA的审评负担。1992年以来,美国新药申请(New Drug Application,简称NDA)数量始终徘徊在100个左右,但是针对新药申请的补充申请(Supplement to NDA,简称SNDA)的数量一致成上升的趋势(如图一),平均每个新药申请的PAS数量一直维持在4-6个,随着总补充申请数量上升最快的是平均每个新药申请的立即生效补充申请的数量(包括CBE-30和CBE报告),为10个,并呈上升的趋势(见图三)。由此可看出,FDA对申请人所提交的PAS数量的取得了一定的成效,控制了每个新药上市申请的PAS的数量,使PAS转化为CBE-30或CBE。
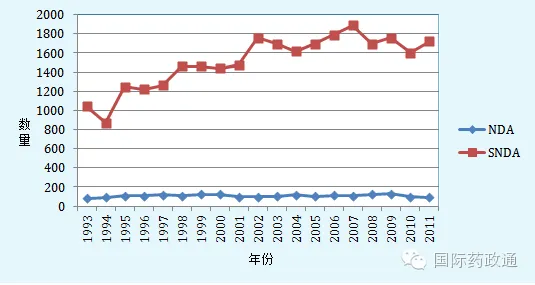
图2 1993-2011年美国新药申请和针对新药申请的补充申请的数量[11]
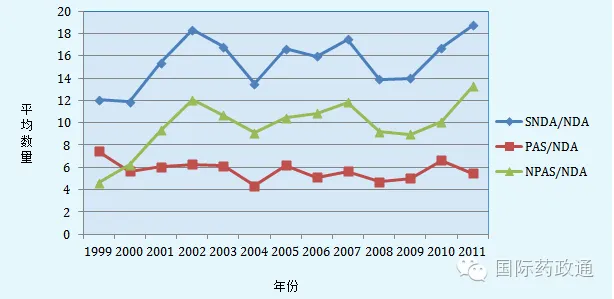
图3 1999-2011年平均每个NDA的PAS以及NPAS(CBE-30和CBE)数量[11]
进行地区试验,验证相关生产变更法规的成本效益
FDA对生产变更的规定详细而又复杂,面对如此繁杂的规定,如何证明其合理性?它给企业政府造成了多大的成本和收益?因此,必须经过合理的验证来支撑所制定法规的合理性。
美国有三部法律分别从政府、企业以及第三方的角度分析所制定的法规合理性。这三部法律分别是Executive Order 12866 、the Regulatory Flexibility Act (5 U.S.C. 601–612), 以及the Unfunded Mandates Reform Act of 1995 (Public Law 104–4) 。 Executive Order 12866指导FDA对可供选择的法规进行成本和效益评估(包括潜在的经济性、对环境的影响、对公众健康和安全性的影响等优势;对发布的影响;公正性),并选择可使效益最大化的立法方法(regulatory approaches )。The Regulatory Flexibility Act 法案规定,拟定的法规必须最小化对大量小实体产生的负面经济性影响。The Unfunded Mandates Reform Act法案第202章节规定,拟定法规的机构必须提交一份预测该法规在整体上对州、地方和部落政府或个体部门每年支出1亿美元的成本和效益的书面评估报告。
基于以上三部法案的理念,美国国会选择运用风险评估的方法制定FDCA法案506A章节,力求降低提交生产变更补充申请对制药行业产生的影响,允许尽早实行生产变更并使药品上市,从而减轻申请人的负担。为了验证该法规对生产企业的影响,在90年代,FDA曾委托外部机构做过一项关于生产变更指南SUPAC–IR 对生产企业的影响的研究,研究发现该指南可减少PAS报告的提交数量,节省生产企业的资源,增加生产企业对于生产资源的控制。研究估计该指南的实施在1997年为生产企业节省将近7100万美元[2]。
美国化学药品生产地点变更制度
对中国的启示
采用风险评估方法划分生产变更,有效分配监管资源,减轻监管机构负担
目前,我国生产变更制度不利于国家相关监管机构有效识别重点监管区域,不能有效灵活监管生产变更。因此,对生产变更的划分应采用风险评估的划分方法,重点监管高风险的生产变更,有效分配监管资源。首先,把风险评估的方法上升到法律的地位,从立法上赋予国家食品药品监督管理总局全程采用风险评估的权力,加强生产变更监管的灵活性,灵活应对风险级别升高或降低的生产变更,建立高风险高监管,低风险低监管的理念,有效利用有限的审评资源,降低企业与监管机构的负担。
减少对场地变更的限制,配合上市许可制度的实行
《药品委托生产监督管理规定》规定境内企业间的委托生产仅限于具有药品批准文号的委托企业才能进行委托,委托企业和受托企业均应是取得相应cGMP的生产企业,并且只有在委托方“因技术改造暂不具备生产条件和能力或产能不足暂不能保障市场供应的情况下,将其持有药品批准文号的药品委托其他药品生产企业全部生产的行为,不包括部分工序的委托加工行为。”该配套的委托规定不仅限制了非生产企业的委托生产,还对生产企业间的委托商业行为进行了限制性规定。并且,《药品委托生产监督管理规定》中还规定“原料药不得委托生产”。
对生产场地变更的限制不利于国家即将逐步实施的创新药上市许可制度(MAH制度)的实行[14]。实施MAH制度的目的是为了改变目前国内药品上市许可与生产许可“捆绑”的现状,允许非生产企业与生产企业自由委托生产[15],使委托生产回归市场化,促使资源的合理配置,建立该制度后,多数创新药生产企业或研究机构必然会将自身生产不经济的生产改为委托生产,然而,国家对药品生产场地变更的限制将会限制该制度的顺利实行。因此,可借鉴美国对药品生产场地变更的规定,扩大生产变更范围,减少对生产场地变更的控制。
结论
化学药品生产场地变更复杂多样,其对药品质量的影响风险程度不同,不能同一而语,必须区分风险高低,加强对高风险的生产地点变更的监管,合理分配有限的监管资源;另外,必须保持相关法规的先进性,例如根据生产技术创新调整生产变更的报告类型,从而增强政府与企业的灵活性,减轻相关政府机构与生产企业的负担,保证药品的安全有效。美国经验值得借鉴,但在后续制度设计方面应充分考虑国情。
REFERENCES:
[1] Federal Register. Supplements and Other Changes to an Approved Application [EB/OL]. [1999-7-28] [2015-05-17]http://www.gpo.gov/fdsys/pkg/FR-1999-06-28/pdf/99-16191.pdf
[6] FEDERAL FOOD, DRUG, AND COSMETIC ACT CHAPTER 9 356a [EB/OL]. [2014-1-1] [2015-05-17]http://www.gpo.gov/fdsys/pkg/USCODE-2013-title21/pdf/USCODE-2013-title21-chap9.pdf
[7]CODE OF FEDERAL REGULATIONS TITLE 21 320.21(c)(1)[EB/OL] .[2014-1-1][2015-05-17]http://www.gpo.gov/fdsys/pkg/CFR-2014-title21-vol5/pdf/CFR-2014-title21-vol5.pdf
[8] FDA. Guidance for Industry CMC Post-approval Manufacturing Changes To Be Documented in Annual Reports [EB/OL]. [2014-3] [2015-05-17]http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM217043.pdf
[11] FDA. FY 1995 - FY 2012 PDUFA Performance Report[EB/OL] [2015-05-17]http://www.fda.gov/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/PerformanceReports/ucm2007449.htm
[12]The first seminar of the working group in CDE. Questions and Answers on drug changes and supplemental application(sessionI)[EB/OL][2009-5-19][2015-05-17]http://www.cde.org.cn/dzkw.do?method=largePage&id=311147
[13]State Food and Drug Administration Administrative Service Center. Audit on Domestic Drug Supplemental Application[EB/OL] [2006-2-20] [2015-05-17]http://www.sda.gov.cn/WS01/CL0372/24058.html
[14]Dandelion.The latest trends and new requirements of drug registration in China from the "National ConferenceforDrugRegistrationin2015"[EB/OL].[2015-4-09][2015-05-17]http://mp.weixin.qq.com/s?__biz=MzAwNzAzMTQ4Mw==&mid=205810497&idx=4&sn=b2984182509dfb652b94870bdd8adb26#rd
[15]Li Xiao-yu , YANG Yue, Liu Qing-jie, et al. Research on Drug Marketing Authorization System Design Based on Revision of the Drug AdministrationLaw[J].Chin Pharm J(中国药学杂志).2015,17:1558-1562
文章原载于中国药学杂志,2016,51(8):671-677,感谢作者惠寄电子版文稿并授权推送,转载此版本请保留出处,并注明转自“国际药政通”(SYPHU-IFDPL)微信公众号。


 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033






 发表于 2017-6-28 16:29:02
发表于 2017-6-28 16:29:02
 置顶卡
置顶卡 变色卡
变色卡

