欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
本帖最后由 孙艳红 于 2025-3-12 16:37 编辑
在制药行业中,无尘洁净环境对于医药企业的生产至关重要。尤其是针对无菌生产产品,其生产过程如果受到污染危害性更大,因此建立符合GMP标准的洁净室(净化车间)显得尤为重要。鉴于产品特性和要求的不同,洁净室的级别和标准也各异。本期gempex德恩咨询专家将深入解析GMP洁净室的级别划分、法规要求和监管重点。

01 洁净室的定义
洁净室,又被称为无尘室或清净室,是指将一定空间范围内之空气中的微粒子、有害空气、细菌等之污染物排除,并将室内之温度、洁净度、室内压力/压差、气流速度与气流分布、噪音振动及照明、静电控制在某一需求范围内,而所给予特别设计的房间。GMP洁净室‌是指满足GMP标准,用于药品生产的洁净室。
02 洁净室级别的由来与划分
洁净室(区)的洁净度级别最早是根据美国联邦标准FED-STD-209E划分为:1级(Class 1)、10级(Class 10)、100级(Class 100)、1,000级(Class 1,000)、10,000级(Class 10,000)和100,000级(Class 100,000),首次提出了洁净室分级的概念,并使用了百级、万级、十万级等分级标准。然而,该标准已于2001年11月29日正式废止,并被ISO 14644标准取代。
目前,ISO14644-1是洁净室洁净度分级的主要参考,它按照空气中颗粒物浓度的不同,将洁净室划分1~9级。在GMP环境下,不同等级的洁净室适用于不同类型的生产和操作过程,例如5~8级适用于无菌产品和其他需要控制悬浮粒子的制药生产。
中国的洁净室标准经历了从模仿到自主创新的过程。早期采用类似于美国FS 209E的标准,后来逐渐过渡到以ISO 14644为基础,并结合本国实际情况进行适当调整。中国新版GMP将洁净室分为A、B、C、D四个等级,每个级别定义了≥0.5μm和≥5.0μm粒子数量的限度,前三个级别分别设定了“静态”和“动态”的限度,D级目前设定了“静态”的限度。
A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域。 B级:A级区的背景区域,即无菌配制和灌装等高风险操作所在的周边环境 C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区,或者非无菌药品生产的环境。
欧盟和其他发达国家也根据自身国情和发展需求,参照ISO 14644-1制定或更新了各自的洁净室标准。
中国GMP法规与其他国家法规对于洁净级别分级的对应关系如下表:
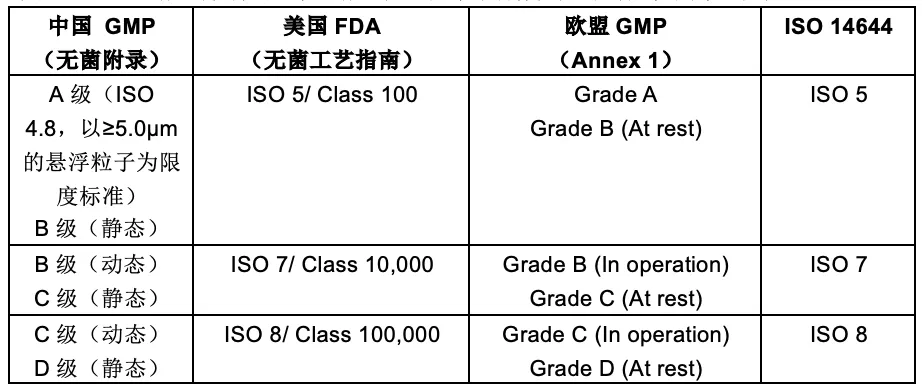
注: 行业内对洁净室的洁净级别分级一般以静态条件下粒子数为参考,如:十万级(Class 100,000)指D级。
03 GMP洁净室监管重点
GMP洁净室作为药品生产的核心区域,扮演着至关重要的角色。它不仅承担着维持极低微粒和微生物污染水平的重任,还需精确调控温度、湿度、压差、照度及噪声等多项环境参数,以防止药品生产受到污染。基于对国内外GMP法规的深入研究,我们的专家总结了7大关键监管重点,旨在帮助制药企业深入理解并实施这些关键要求,以建立一个既符合GMP标准又能满足生产需求的洁净环境。
1. 悬浮粒子
洁净区的设计必须符合相应洁净度级别的要求,中美欧对于各级洁净区悬浮粒子的要求有所不同,但均对“静态”和“动态”条件下的粒子数进行了详细规定。
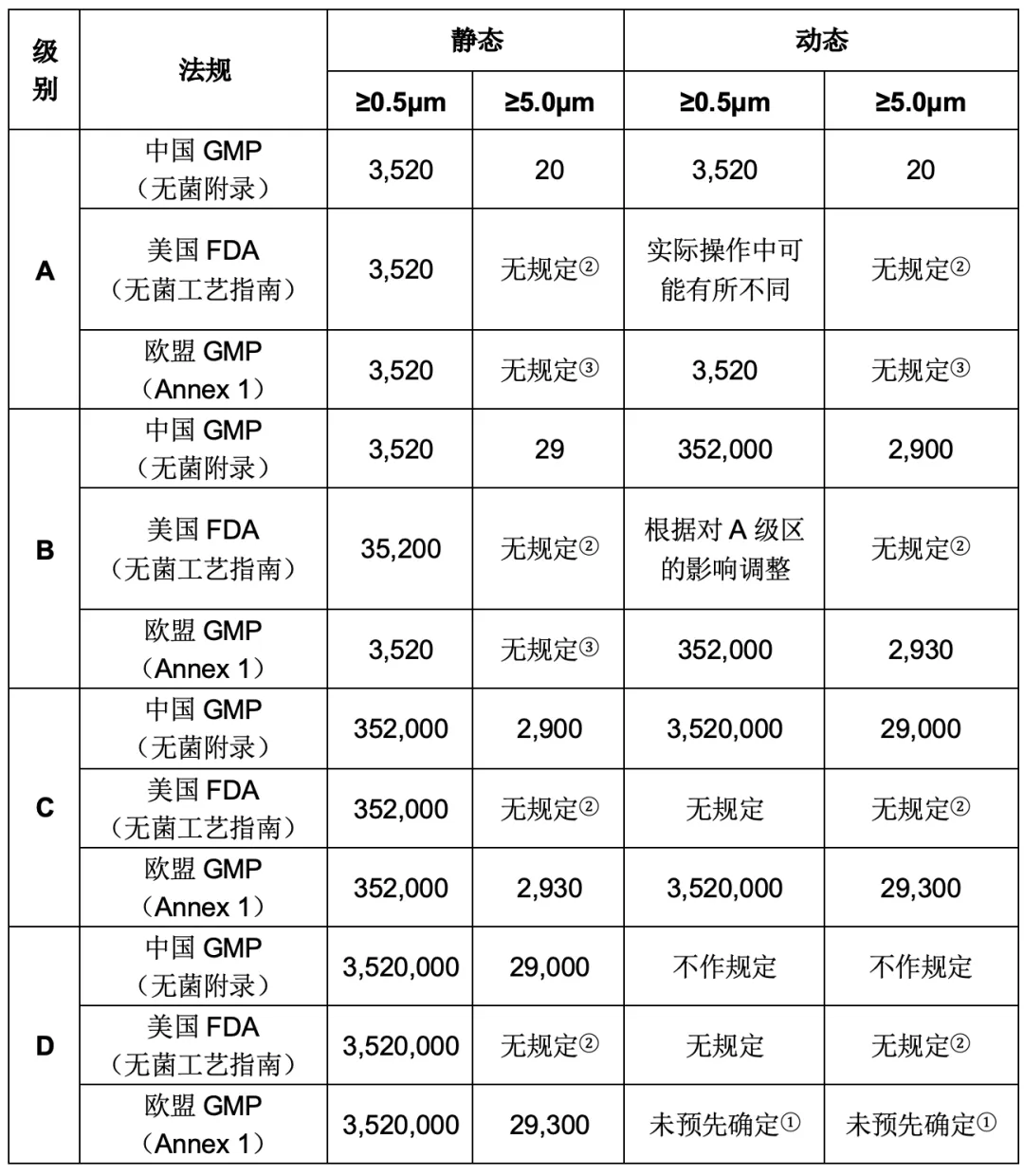
注: ① 对于D级,动态限度未预先确认。生产商应根据风险评估和适用的常规数据建立动态限度; ② FDA指南未对≥5.0μm悬浮粒子的最大允许数进行规定,可参考ISO14644-1进行规定; ③ 标准可根据历史数据和CCS来制定。
可按照质量风险管理的原则对洁净室(必要时)进行动态监测。监控要求以及警戒限度和纠编限度可根据操作的性质确定,但自净时间应当达到规定要求,即:生产操作全部结束、操作人员撤出生产现场并经15~20分钟(指导值,欧盟要求只需要小于20分钟)自净后,洁净区的悬浮粒子达到“静态”标准。
2. 微生物
在微生物限度方面,中国GMP和美国FDA无菌工艺指南主要规定在动态环境监测中进行微生物监测,而EU GMP 附录1则要求在洁净室确认(静态)以及环境监测(动态)中均需要进行微生物监测。
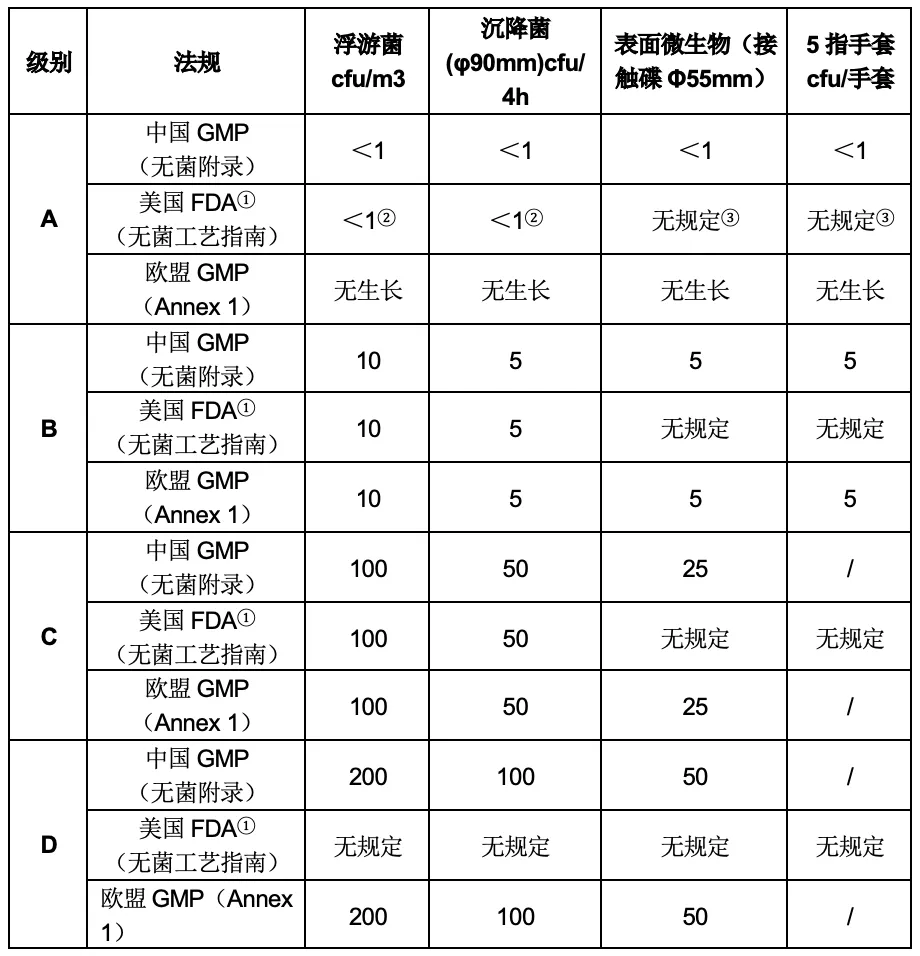
注: ① FDA的无菌工艺指南中推荐的纠编限对应ISO 8级别; ② 在ISO 5环境中取样通常不得有微生物生长; ③ 可进一步参考USP<1116>。
3. 温度和湿度
目前GMP没有对温湿度作出强制的具体值标准。企业应从产品工艺风险出发,考虑相应的国家标准/行业标准和技术指南制定相应的温湿度标准。比如在《医药工业洁净厂房设计标准》GB 50457-2019中写道:
4. 压差
为保证洁净区的空气洁净度不会受到污染空气的干扰,不同级别洁净区之间以及洁净区与非洁净区之间需保持一定的压差。中美欧对于洁净区压差的要求基本一致。

注: ① FDA无菌工艺指南原文提到的是无菌处理房间与相邻的未定级房间之间应至少保证有12.5的压差。建议读者可以延伸阅读。
必要时,相同洁净度级别的不同功能区域(操作间)之间应保持适当的压差梯度。(如:5 Pa)
5. 照度和噪声
目前GMP没有对照度和噪声没有作出强制的具体值标准。
企业应从产品工艺风险以及人员舒适度/职业防护等方面出发出发,考虑相应的国家标准/行业标准和技术指南制定相应的标准。比如在《医药工业洁净厂房设计标准》GB 50457-2019中写道,主要工作室一般照明的照度值宜为300lx;辅助工作室、走廊、气锁、人员净化和物料净化用室的照度值宜为200lx。非单向流医药洁净室的噪声级(空态)不应大于60dB(A),单向流和混合流医药洁净室的噪声级(空态)不应大于65dB(A)。
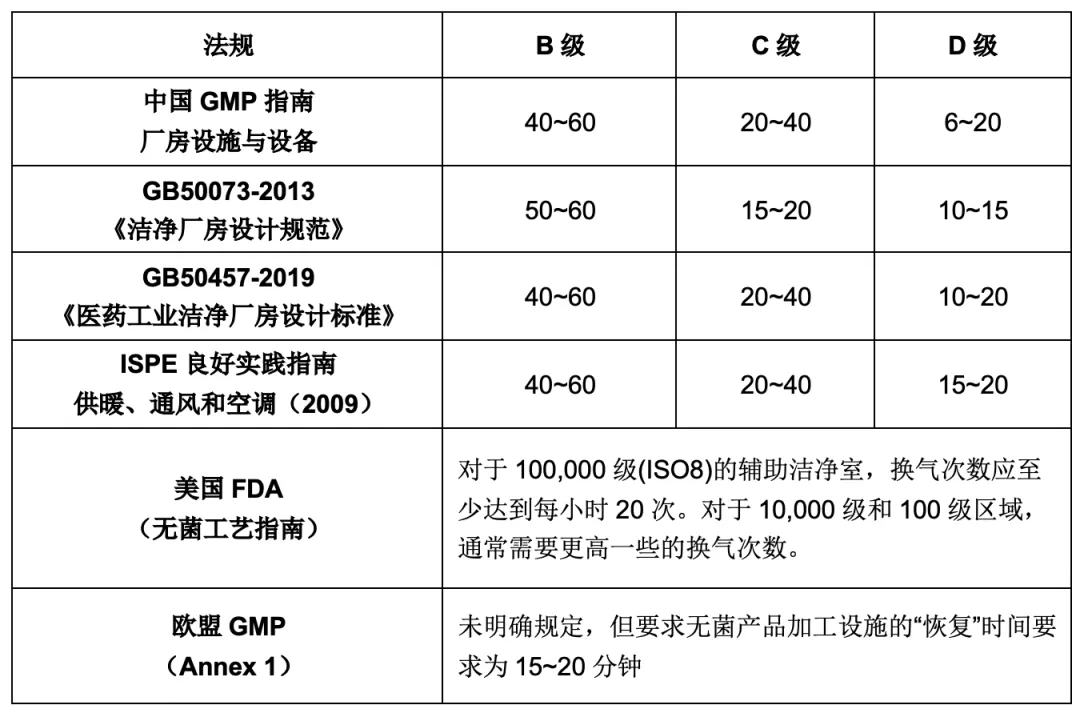
6. 换气
许多GMP法规和指南没有明确规定换气量,而是由项目设计者来分析和定义。然而,通过对各国GMP法规和指南的研究发现,GMP规定了洁净室的换气次数指导值为每小时换气20次 (ACH),自净(也称为恢复)时间指导值为15~20分钟。洁净室从使用状态到静止状态的恢复过程与其换气次数直接相关,换气次数越高,恢复过程越快。
7. 人员控制
中国、美国和欧盟GMP法规对洁净室人员控制的要求在核心原则上保持一致,均强调人员数量控制、人员培训、个人卫生、着装防护和行为约束等方面。然而,在具体培训内容、健康检查要求、着装与防护细节以及行为约束措施等方面,各国GMP法规可能存在一定差异。
在人员数量控制上,中美欧GMP都强调要严格控制进入洁净区的人数,以减少人员活动带来的污染风险。各相关洁净区可考虑设计门禁系统,限制无权限人员进出,并对进出人数进行控制。洁净区不同房间最大允许进入人数可按照以下的方式进行计算,取计算的最小值确定洁净区房间最大允许进入人数: - 按照洁净区内新鲜空气量进行计算;
- 根据不同的空气洁净度等级和工作人员数量进行计算。
综上所述,GMP洁净室的标准与建立涉及多个方面,需综合考虑产品特性、生产工艺及各国GMP法规的要求。通过深入分析中美欧等国的GMP法规,我们可以更全面地了解洁净室悬浮粒子、微生物、温湿度、压差、照度、噪声、换气及人员控制等方面的具体要求,从而确保医药产品的生产环境满足GMP标准。
P.S. 本文章为gempex德恩咨询原创。如需转载,请注明来源于gempex德恩咨询。
关于gempex德恩咨询 德恩咨询是gempex在中国的全资子公司,是具有国际影响力的GMP咨询与执行机构,致力于为全球的生命科学企业提供合规、高效及可执行的GMP解决方案。经过22年的发展,我们拥有60多位经验丰富的GMP专家,全球累计执行项目超过5000个,累计为1000多个客户提供专业服务,业务遍布20多个国家,并与众多知名药企建立了长期的合作关系。 我们的专家团队拥有丰富的行业经验,熟知NMPA、FDA、EU、WHO、ICH、PIC/S、MHRA、SWISSMEDIC、TGA等GMP法规要求,能为不同国家和地区的客户提供定制化的解决方案,服务包括全球GMP符合性、新厂房合规性、CS计算机化系统、工厂质量管理和多国MAH/MAA服务。
| 

 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033






 发表于 2025-1-20 17:10:34
发表于 2025-1-20 17:10:34
 置顶卡
置顶卡 变色卡
变色卡
