欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
对一个医疗器械从业人员来说,FDA网站绝对是一个宝藏库。比如在产品开发时搜索在美国上市的同类产品的技术申报资料的摘要,类似于目前中国国家药品监督管理局发布的产品《医疗器械产品注册技术审评报告》。区别在于,目前中国NMPA仅就个别产品发布该报告,而FDA将所有通过510(k), PMA和De Novo的产品都公开相关资料,经PMA路径审批的产品还会公开说明书和标签。
1、 FDA医疗器械的分类
FDA对医疗器械实行分类管理,根据风险等级和管理程度,分为三类(I,II,III)进行上市前管理。
I类产品为“普通管理(General controls)”产品,FDA对这类产品大多豁免了上市前通告程序(Pre-market Notification,PMN;即510(k)),通过登记管理,即可上市。
II类产品为“普通&特殊管理(General & Special Controls)”产品,这类产品基本上都需要进行510(k),才可上市销售。有极少数的一部分II类产品可以豁免510(k)。
III类产品为上市前批准管理(Pre-market Approval,PMA)产品,生产企业在产品上市前必须提交PMA申请和相关资料,证明产品符合要求。
除了以上三类外,全新的无已上市同类/相似产品的医疗设备则通过“De Novo”申请;进行分类判断并确定需要递交哪些材料。
2、如何检索FDA的数据库 A)通过医疗器械数据库检索 我们登陆FDA医疗器械部分,链接https://www.fda.gov/Medical-Devices找到数据库,进入产品对应的数据库搜索相关产品。  还有一个更方便的链接, https://www.accessdata.fda.gov/scripts/cdrh/devicesatfda/index.cfm 这个链接包括了510(k)和PMA两个数据库(如下图),基本囊括了绝大多数的IVD产品。 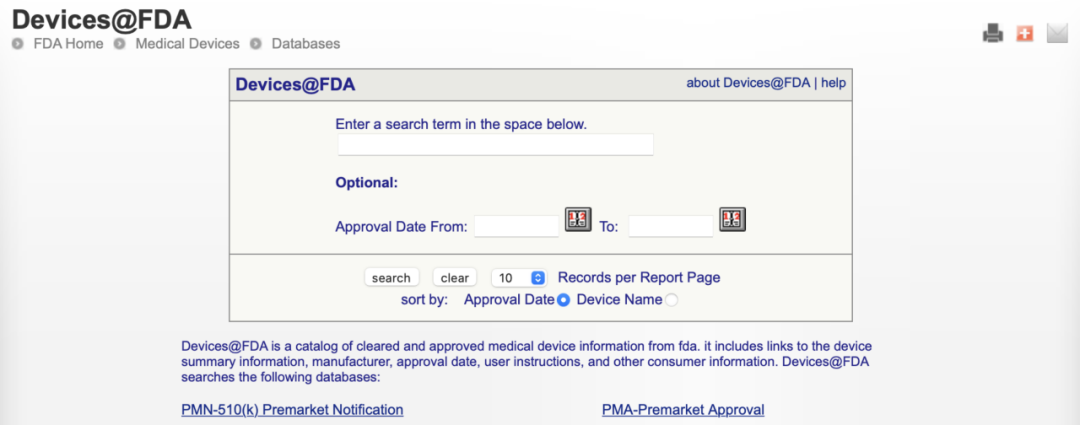 B)通过分类检索
FDA对部分IVD产品还进行了分类总结,如伴随诊断产品,血糖监测产品,分子诊断产品等等,进入对应分类后,可以非常容易查到这个分类下的所有FDA获批的产品。 https://www.fda.gov/medical-devices/products-and-medical-procedures/in-vitro-diagnostics 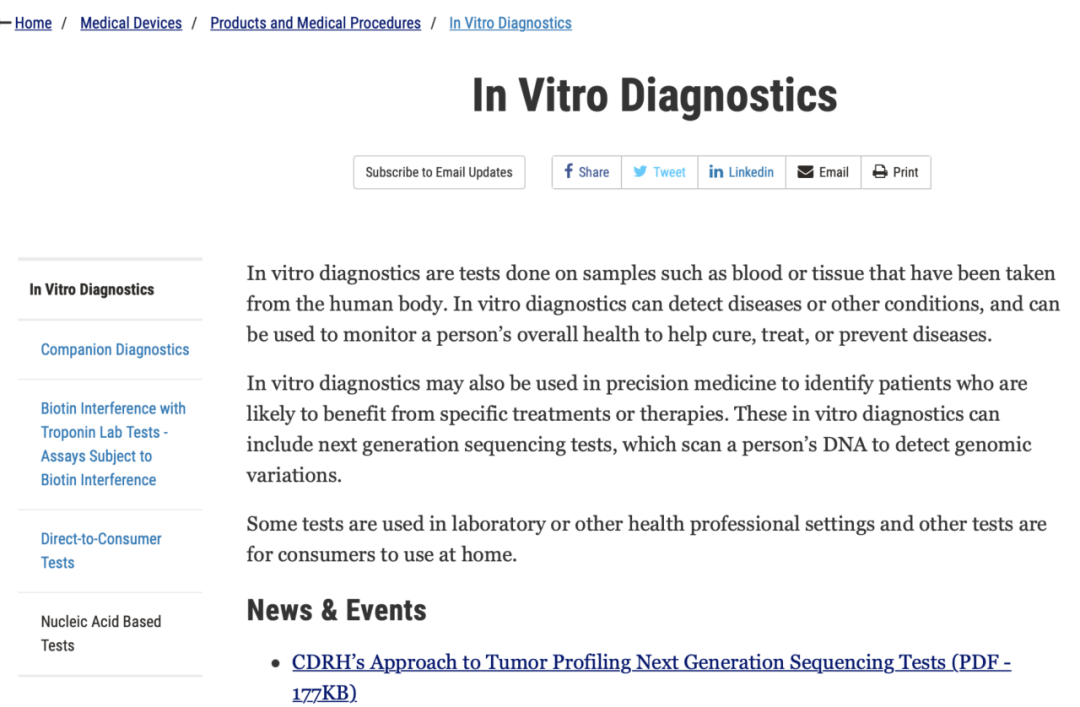
以分子诊断产品举例:点击进入后,可以看到如下的页面,产品进行了第二次分类,人基因检测产品和微生物类检测产品。
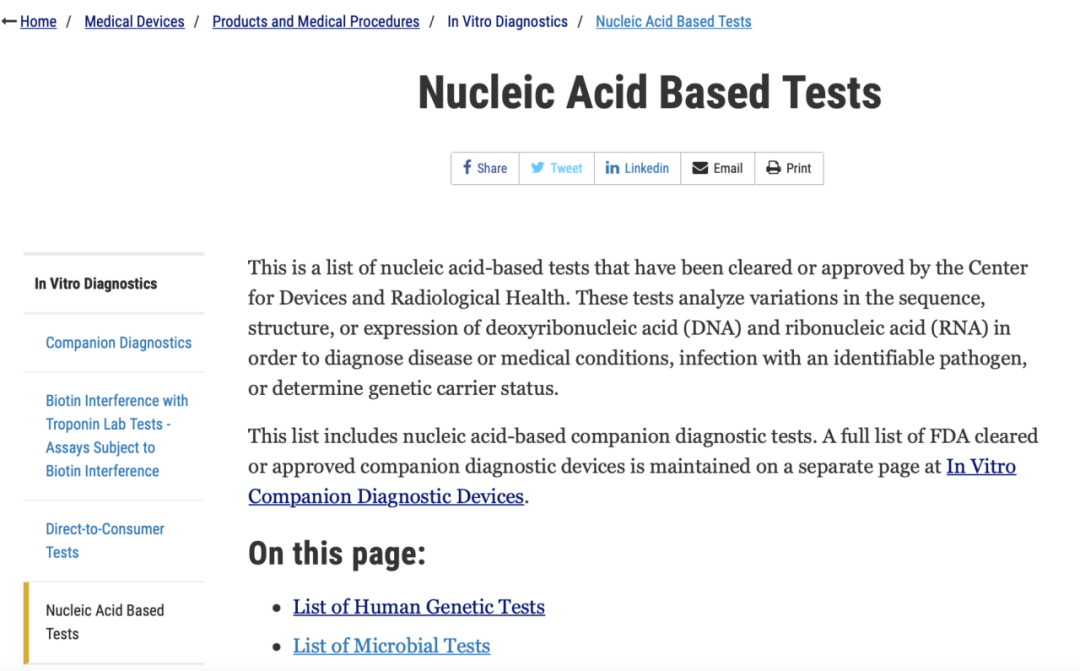 所有产品均按照疾病进行的分类,列明了该疾病所有FDA批准上市的诊断试剂,比如下图就是肝炎检测的相关分子诊断产品。 比如我们想了解美国批准上市的伴随诊断产品的情况,只需要进入“Companion Diagnostics”那一部分,就可以快速的对其有一个全面了解,而无需再一个个去搜索。
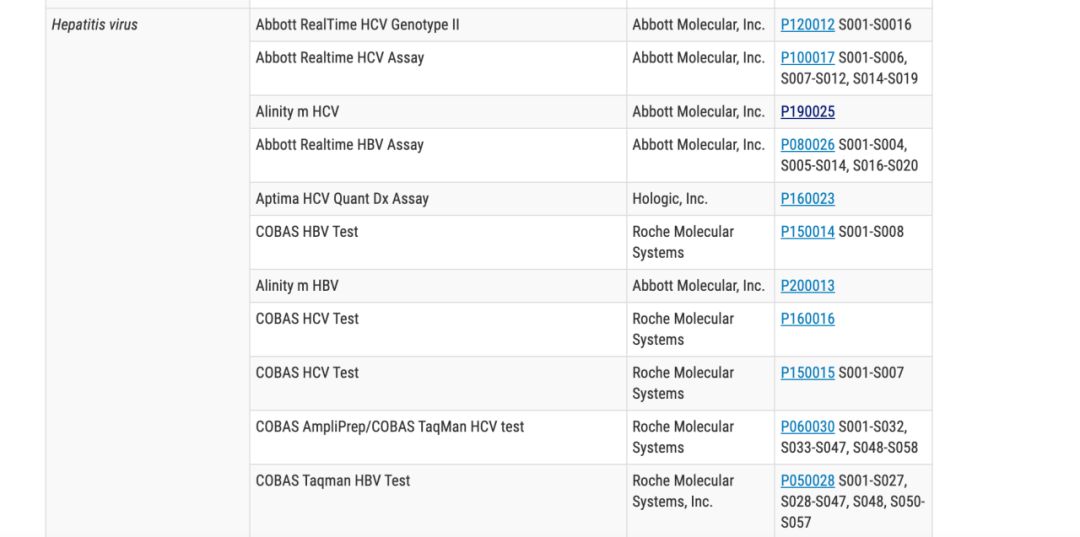 点击后面的510(k) Number 或PMA Number,可以直接链接到数据库,以上图第三个雅培的Alinitiy HCV举例,可以链接到如下界面。 这个界面和通过数据库检索得到的界面是相同的,可以看到,该产品是在2020年第一次获批,并提交了6次补充资料。进入对应的链接,就可以下载到相应的资料了。
| 
 [复制链接]
[复制链接]
 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033






 发表于 2021-7-2 08:53:02
发表于 2021-7-2 08:53:02

 置顶卡
置顶卡 变色卡
变色卡

